Desmosomes in Inherited Disease of Heart and Skin
Desmosome molecules are targets in inherited, autoimmune and bacterial toxin-mediated skin disease. Inherited mutations in desmosome molecules also lead to a potentially lethal sudden death syndrome called arrhythmogenic cardiomyopathy (AC). Sometimes patients have a combined cardiocutaneous disorder cause by mutations in desmosome molecules.
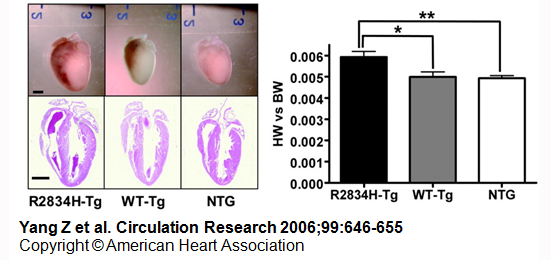
Heart Disease: In vitro biochemical and in vivo strategies are being used to understand how specific human gene mutations result in disease. For instance, we showed that a desmoplakin mutation R2834H, which causes AC in humans and in a transgenic model of the disease (Fig. 1), inhibits a GSK3beta dependent phosphorylation cascade in the DESMOPLAKIN C-terminus by preventing methylation of an upstream arginine, which likely conformationally prepares desmoplakin for proper modification (Albrecht, et al. 2015. J. Cell Biol. 208: 597). Phosphorylation in turn is required for desmoplakin’s normal dynamics and assembly into desmosomes. The amino-terminus of desmoplakin harbors additional functions important in AC. For instance, through a protein interaction screen we identified the microtubule end-binding protein EB1 as a new binding partner for this region. AC mutations that interfere with EB1 binding result in loss of microtubule stability at the plasma membrane (Fig. 2) (Patel, et al, 2014. J. Cell Biol. 206: 779). This in turn impairs the stable incorporation of connexin 43 into gap junctions, and loss of gap junction communication in cardiac myocytes, which could contribute to defects in electrical conduction in AC.
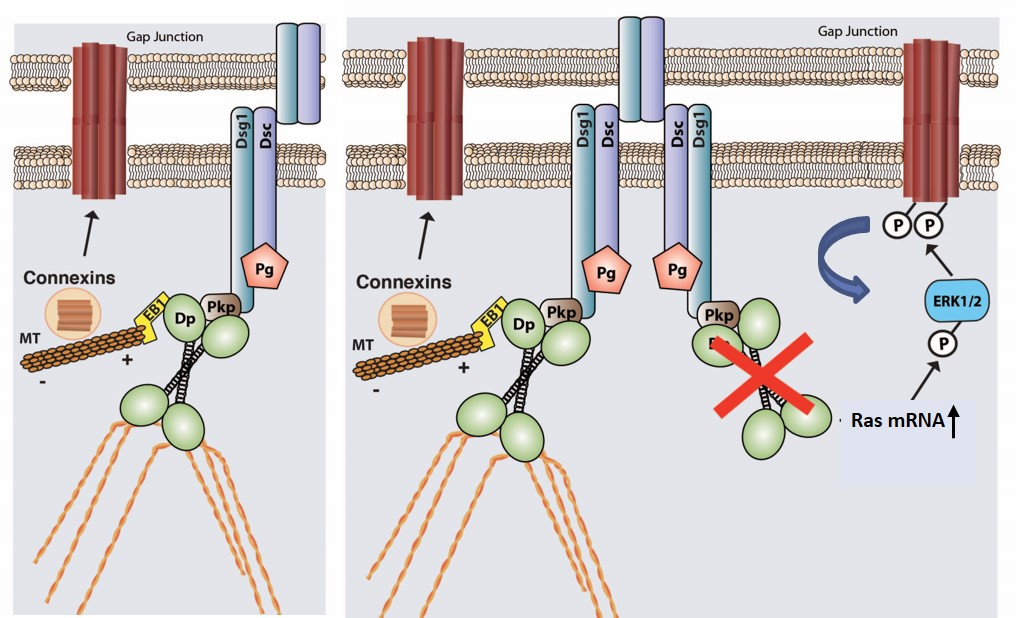
We recently discovered the desmoplakin also regulates connexin 43 stability by dampening Ras-mediated Erk-dependent lysosomal turnover (Fig. 2) (Chen, et al. 2018. J. Cell Biol. 217: 3219). The contributions of PKC and RhoA signaling defects in patients with AC caused by PKP2 and desmoplakin mutations is also being investigated. We collaborate with several labs to address the role of desmosomes in heart, including Drs. Farah Sheikh, Jeff Saffitz, and Mario Delmar in this area, as well as Simona Polo, Paul Lampe, and NU colleague Karla Satchell.
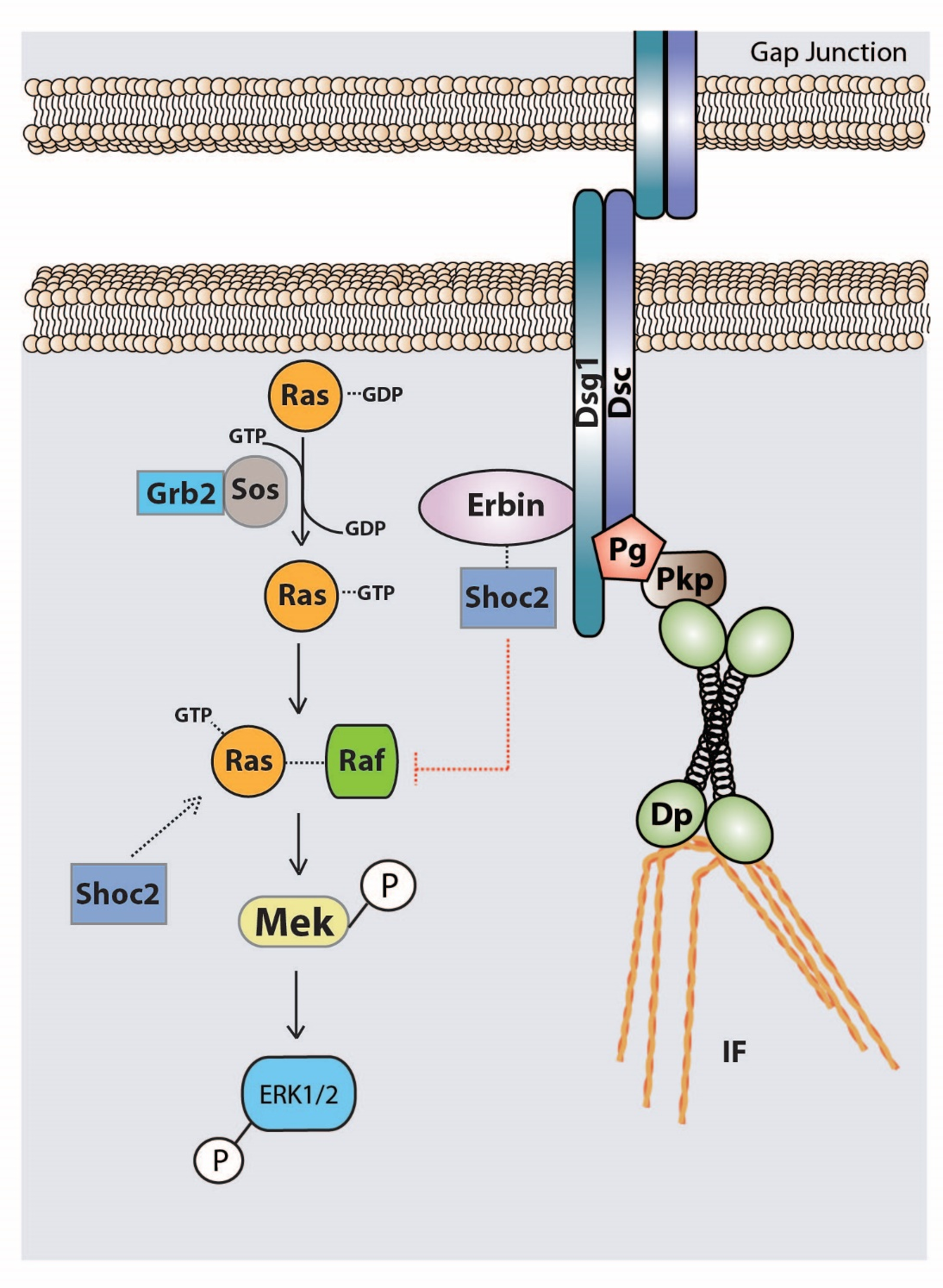
Skin Disease: Inherited mutations in multiple desmosome molecules also cause a variety of skin disorders. We are collaborating with Eli Sprecher at Sourasky Medical Center, Tel Aviv, to understand the underlying basis of these genetic disorders. In the case of desmoglein 1, haploinsufficiency results in striate palmar plantar keratoderma (SPPK) whereas a more complete loss of function results in a severe disorder called SAM syndrome (severe dermatitis, multiple allergies and metabolic wasting syndrome). In both cases, Dsg1 functions that transcend their roles in adhesion may contribute to disease phenotypes. Our understanding of these functions comes from a protein interaction screen for novel binding partners, which uncovered the PDZ domain-containing protein called Erbin as a novel binding partner of the desmoglein 1 cytoplasmic tail. We showed that Erbin couples desmoglein 1 to the Erk1/2 signaling pathway, and is required for desmoglein 1 to suppress MAPK signaling by interfering with Ras-Raf coupling, which involves Shoc2. Inhibition of MAPK by Dsg1 is important for promoting proper keratinocyte differentiation (Harmon, et al., 2013. J. Clin. Invest. 123:1556). Interference with this pathway is accompanied by defects in differentiation and hyperkeratosis in patients with desmoglein haploinsufficiency and SPPK. This pathway also appears to be involved in regulating paracrine signaling in keratinocytes. In a collaborative study with Asma Smahi’s group, loss of Dsg1 in patients with a variety of SAM syndrome (called SAMEC) resulted in the cell autonomous production of cytokines via an NFkB pathway, requiring Erbin (Polivka, et al. , 2018. J. Allerg. Clin. Immunol. 142:702.). How these cytokines contribute to systemic defects observed in patients with this disease and also in cancer (see Desmosomes and Cancer page) is an area of active investigation.
Future Objectives: Future objectives include determining the role of desmosomes in shaping cell mechanics and in transmitting mechanical signals into changes in cell behavior. As one example, we have identified a new interaction between the Rho GEF Ect2 and desmoplakin, which is disrupted in cells from Carvajal patients with desmoplakin mutations. Through Ect2, desmoplakin recruits Rho to cardiac myocyte and keratinocyte cell-cell junctions to regulate cardiac cell mechanics and contraction. Atomic force microscopy, traction force microscopy and other tools to probe cell mechanics are being used to determine the importance of desmoplakin-Ect2 interactions in heart and skin. We are also defining other shared and distinct pathways that occur in patients with cardiac and cutaneous symptoms that influence disease including regulation of the MAPK pathway and connexin-based gap junctions. The importance of directing patients with cutaneous symptoms (e.g. wooly, kinky hair) for cardiac evaluation is being established in collaboration with Dr. David Kelsell’s group in this area.