Developmental Transcription Elongation Control in Disease
Transcriptional Elongation Control in Disease and Development
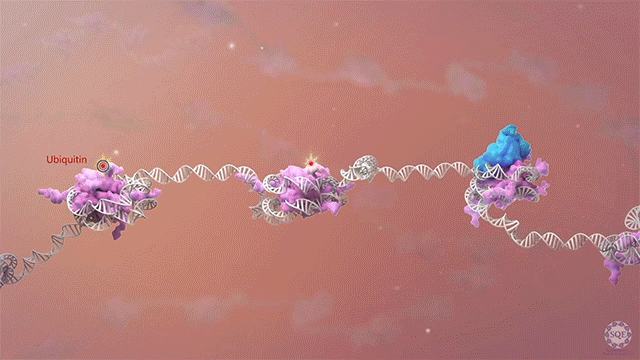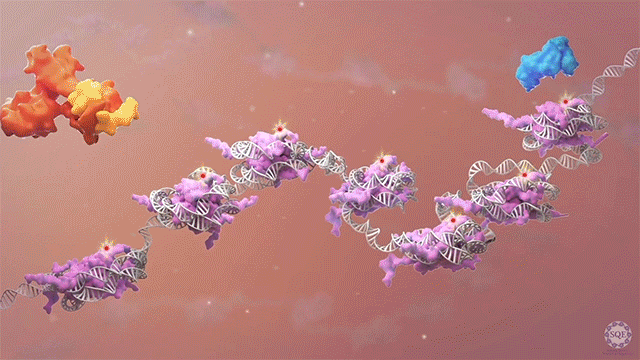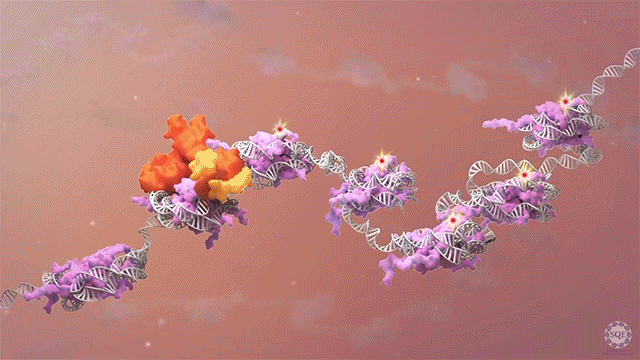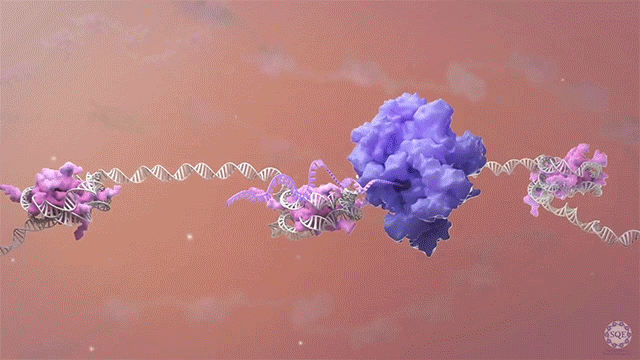
Following transcriptional initiation and escape from promoter regions, RNA Pol II pauses 20-60 bp downstream of the TSS, prior to its release into gene bodies for productive transcript elongation. Previously, biochemical studies implicated several factors in the regulation of this pausing step, including Negative Elongation Factor (NELF) and P-TEFb. However, we recently reported that contrary to its apparent activity in vitro, NELF does not appear to function as a negative regulator of elongation in vivo. Upon auxin-induced NELF depletion, RNA Pol II was not released from promoter-proximal pause sites into gene bodies, but instead accumulated at a second, downstream pause site associated with the +1 nucleosome. Moreover, forced addition of NELF did not impede transcript elongation. However, we did observe a NELF-dependent loss of the cap-binding complex (CBC) from promoter regions, and our analysis indicated that NELF interacts with CBC to regulate the stability and early termination of nascent transcripts. [Aoi et al., 2020]
NELF in RNAPII Pause-Release Video
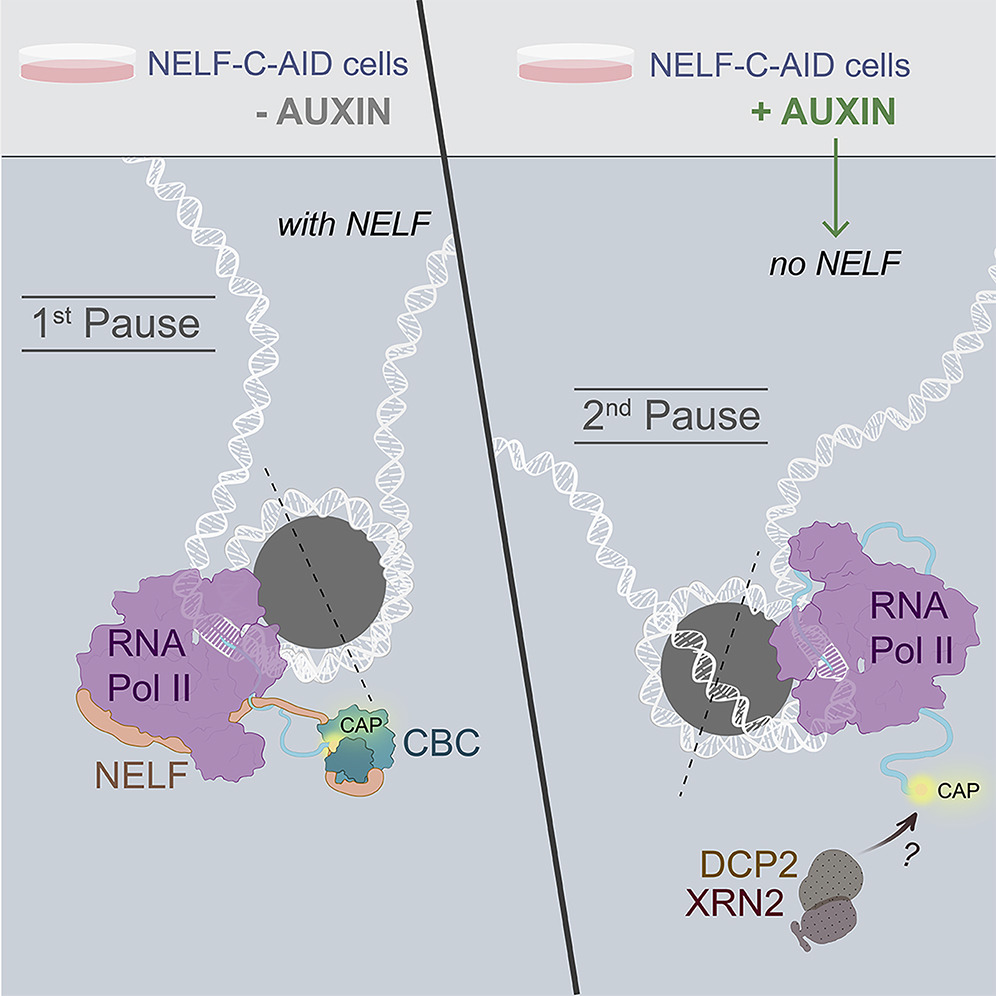
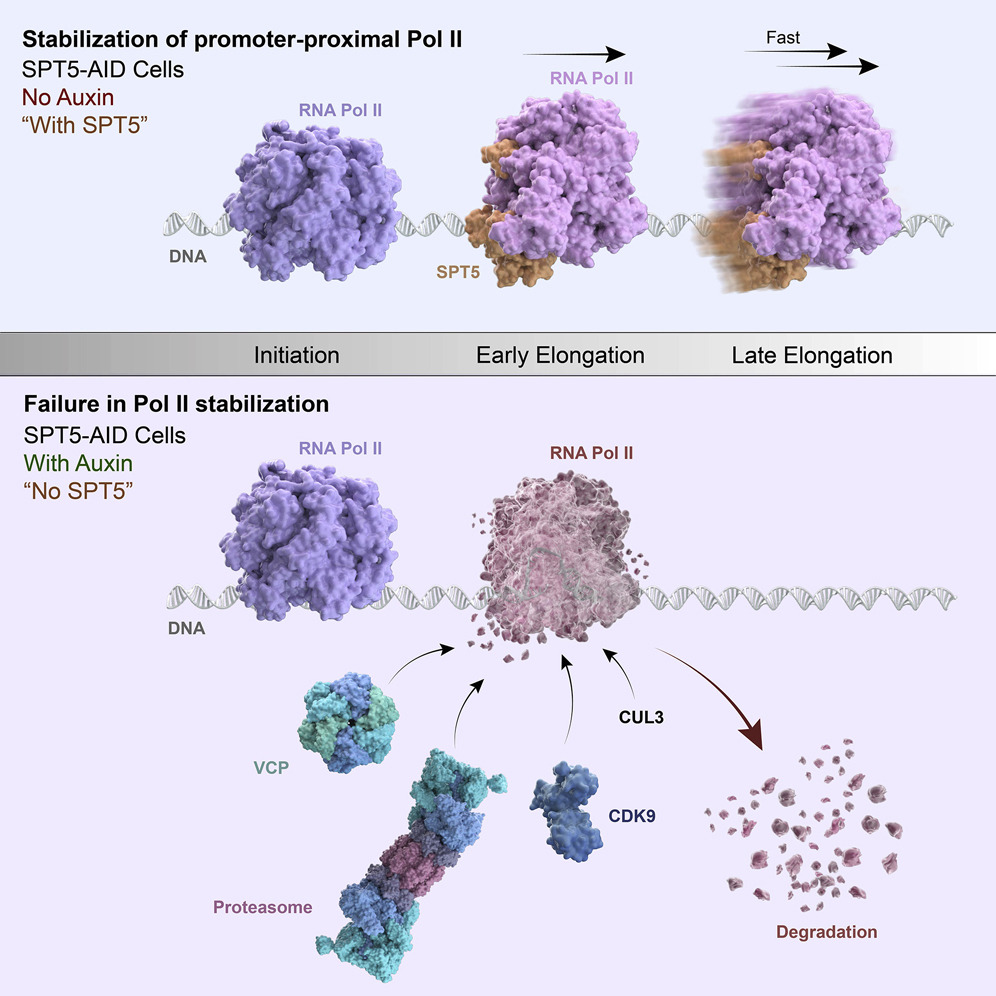
Rapid depletion also revealed that NELF participates in the pause-regulating activity of SPT5, a subunit of the RNA Pol II complex member DSIF. Promoter-proximally paused RNA Pol II was profoundly destabilized upon SPT5 depletion, due to unchecked degradation of the polymerase subunit RBP1 by the unfoldase VCP. By stabilizing RNA Pol II with a VCP inhibitor, we were also able to observe a loss of RNA Pol II-NELF interaction in the absence of SPT5, accompanied by the second-site pausing phenomenon seen upon NELF depletion. Therefore, SPT5 appears to stabilize paused RNA Pol II both by protecting its RBP1 subunit from degradation and by maintaining its association with NELF. [Aoi et al., 2021]
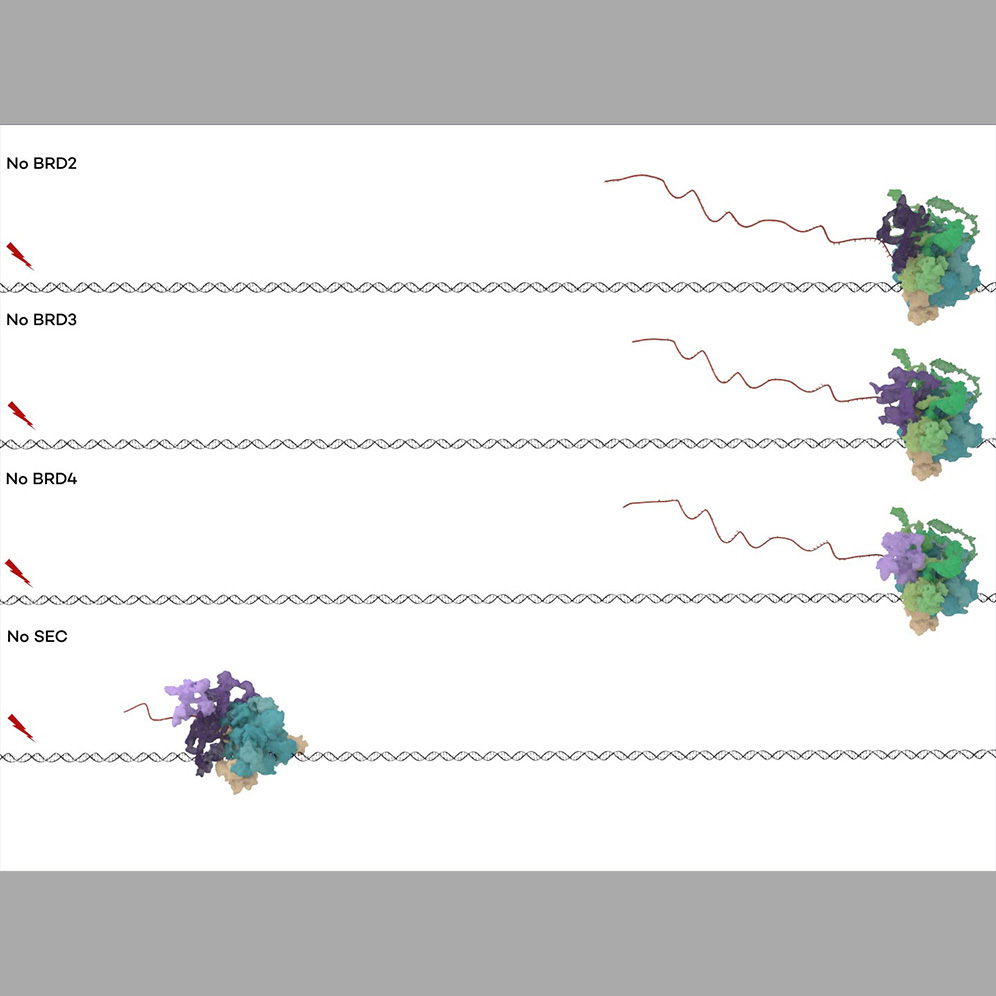
We can leverage rapid depletion strategies to differentiate functional roles and determine causal relationships between elongation factors and complex members. For example, Cdk9/P-TEFb can form a catalytically active, transcription-regulating complexes both with the bromodomain-containing BET family member BRD4 and the Super Elongation Complex (SEC). Additional BET family members also regulate transcription, and the SEC is itself variably composed of alternative complex members. We therefore made use of orthogonal depletion strategies to distinguish the functional roles of BET and SEC protein family members. We generated separate AID cell lines for auxin-induced depletion of each BET family member, and also used the JQ1-based PROTAC dBET6 to deplete all four members simultaneously in the parental cell line. Comparing the effect of these depletions on RNA Pol II behavior, we found that BET-dependent loss of promoter-proximal pause release under normal conditions was only recapitulated by depletion of BRD4, while only BRD2 depletion could recapitulate the BET-dependent loss of RNA Pol II from distal enhancers. BET depletion did not affect pause release in response to heat shock. However, using AID to deplete the alternative SEC member scaffold proteins AFF1 and AFF4, we found that loss of either member disrupted promoter-proximal pause release in response to heat shock, with AFF4 depletion having a much stronger effect. Taken together, these findings indicate that BRD2 controls RNA Pol II activity at enhancers, that the BRD4-PTEFb complex controls release of promoter-proximal paused RNA Pol II under normal conditions, and that the AFF4-containing form of SEC primarily controls transcriptional activation in heat shock response. [Zheng et al., 2021]
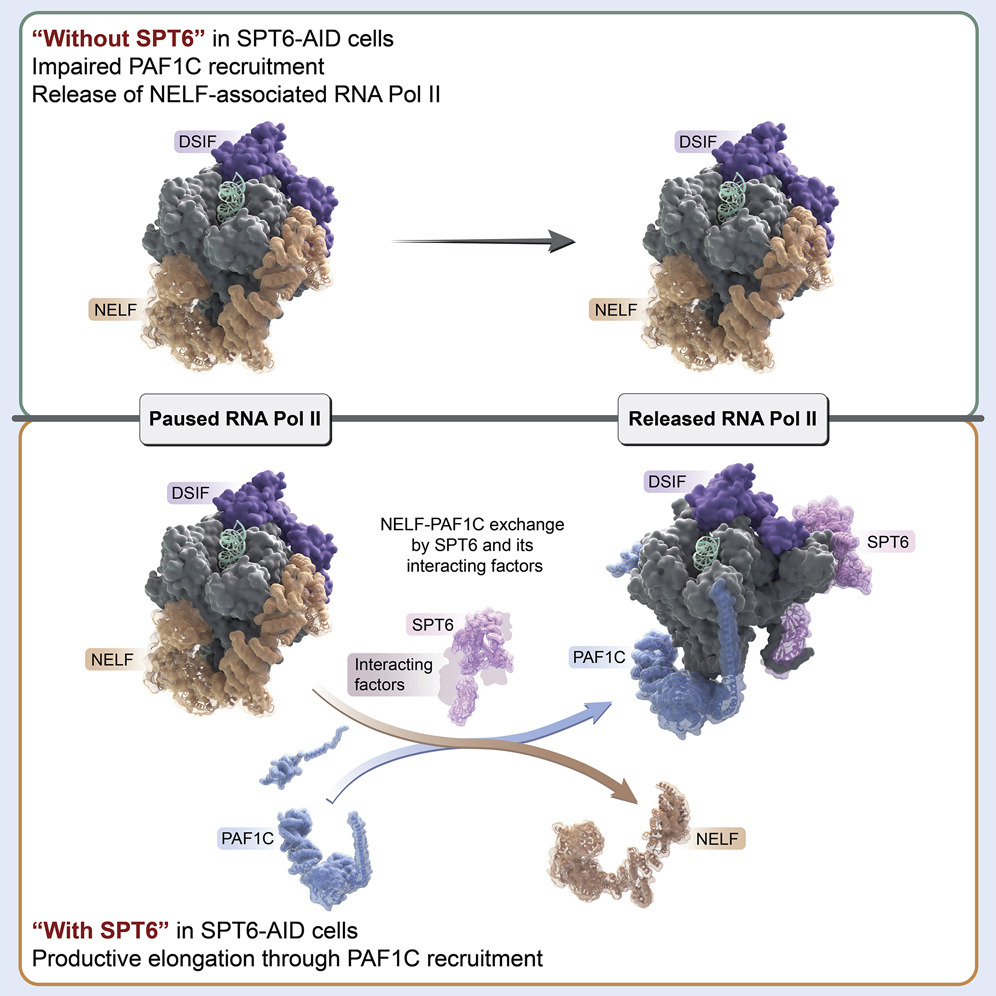
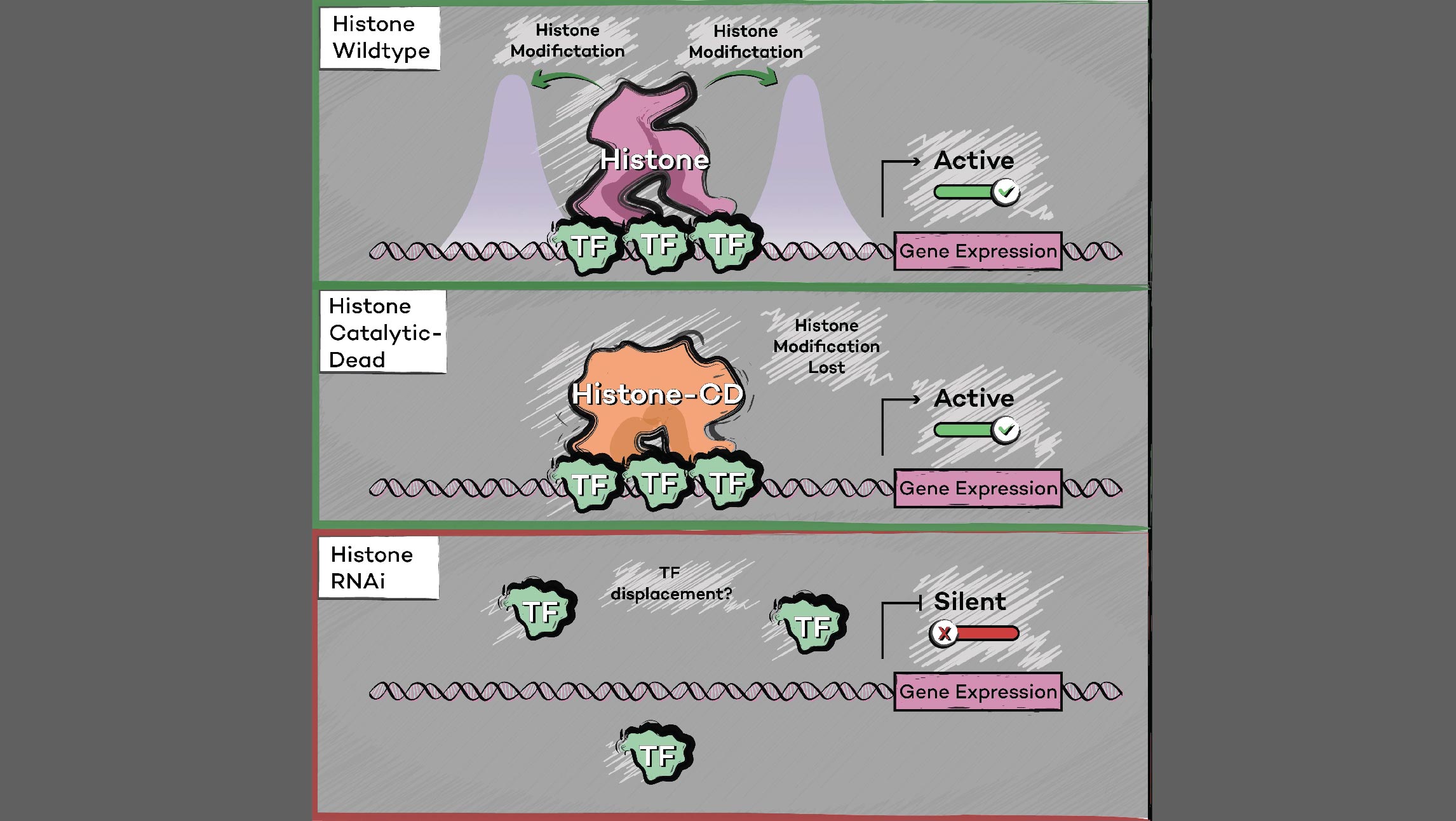
We previously reported that the PAF1 complex (PAF1C) interacts with RNA Pol II to regulate pause-release [Chen et al., 2017]. Like PAF1C, the elongation factor SPT6 also regulates co-transcriptional histone modifications, and the causal relationship between these factors was initially unclear: we observed similar release of RNA Pol II into gene bodies, reduced processivity and defective heat shock-induced pausing upon depletion of either PAF1C or SPT6. However, we found that while PAF1C depletion had no effect on the interaction between RNA Pol II and SPT6, conversely, SPT6 depletion disrupted interactions of RNA Pol II not only with PAF1C but also with other transcript processing factors. In addition, the interaction of RNA Pol II with NELF was also stabilized upon SPT6 depletion, and this interaction aberrantly persisted into gene bodies. This resolution of the causal relationship between SPT6 and PAF1C allowed us to identify new functional roles for SPT6 in elongation control: 1) controlling pause release by direct interaction with the RNA Pol II complex, and 2) licensing productive elongation by recruiting PAF1C to replace NELF on paused RNA Pol II. [Aoi et al., 2022]
Though not directly applicable to clinical medicine, rapid depletion strategies are powerful research tools that can support the development of new disease therapies. For example, because depletion of PAF1C is known to increase viral transcription in HIV-1-infected cells, we recently sought to identify a small molecule inhibitor that could impair PAF1C chromatin recruitment. To do so, we first used in silico molecular docking simulations to identify small molecules that could inhibit PAF1C formation by blocking physical interaction between the subunits PAF1 and CTR9. Then, we evaluated the efficacy of our candidate molecules by assessing their effects on Pol II pausing in vivo. Our approach yielded an effective small molecule inhibitor, iPAF1C, which we further validated using rapid depletion of PAF1C as a positive control. Finally, we were able to demonstrate not only that iPAF1C enhanced the viral transcription-inducing activity of latency reversal agents (LRAs) in cell line latency models, but also that it was sufficient to induce latency reversal on its own in ex vivo cells from HIV-1 patients. [Soliman et al., 2023]