Our Research
We are using genomic approaches to examine why some bacterial strains within a species are more virulent or transmissible than others. These investigations have been applied to multidrug-resistant bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae. To date, studies have led to the identification of several novel virulence factors such as CdiA, which is a contact-dependent inhibition protein that represents a new family of virulence factors. Other studies have identified “high-risk” strains of multidrug resistant bacteria that have spread locally at our hospital or globally. We are expanding these studies to identify additional novel virulence and colonization/transmission factors that will then be characterized using molecular techniques, microscopy, tissue culture and mouse models to explore their mechanisms.

Description: This animation depicts the type III secretion system of P. aeruginosa. Needle-like apparatuses of the T3SS inject toxins directly into the host cell. Four effectors: Exo S/T/U/Y are shown here.
In a second project, we investigate the role of the type III secretion system of P. aeruginosa in pathogenesis. Type III secretion pathways form needle-like apparatuses that inject toxins directly into the cytoplasm of host cells. They are used by many gram-negative bacteria to injure or kill human cells and thus cause disease. We focus on the interaction of the secreted toxins with the host. Current projects involve the use of molecular and biochemical techniques to identify novel type III secretion effector proteins and to examine co-factors that modulate type III secretion phenotypes.
The common theme of these projects is to better understand the mechanisms by which multidrug-resistant bacteria cause disease and spread.
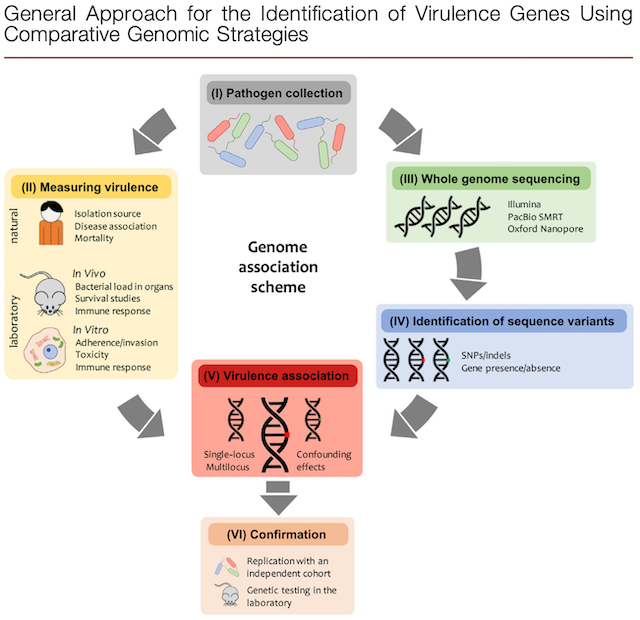
Description: This diagram shows the approach for identifying virulence genes using comparative genomics. This includes: (1) obtaining large amounts of isolates (2) testing the virulence of these isolates (3) whole genome sequencing the isolates (4) mapping the genetic differences (5) associating the genetic differences with the virulence data and (6) confirming the virulence-associated elements with an independent cohort to confirm loss or gain of virulence.