Software & Protocols
scNanoGPSSingle cell nanopore RNAseq data analysis for detecting Genotype-Phenotypes simultaneously in same cells
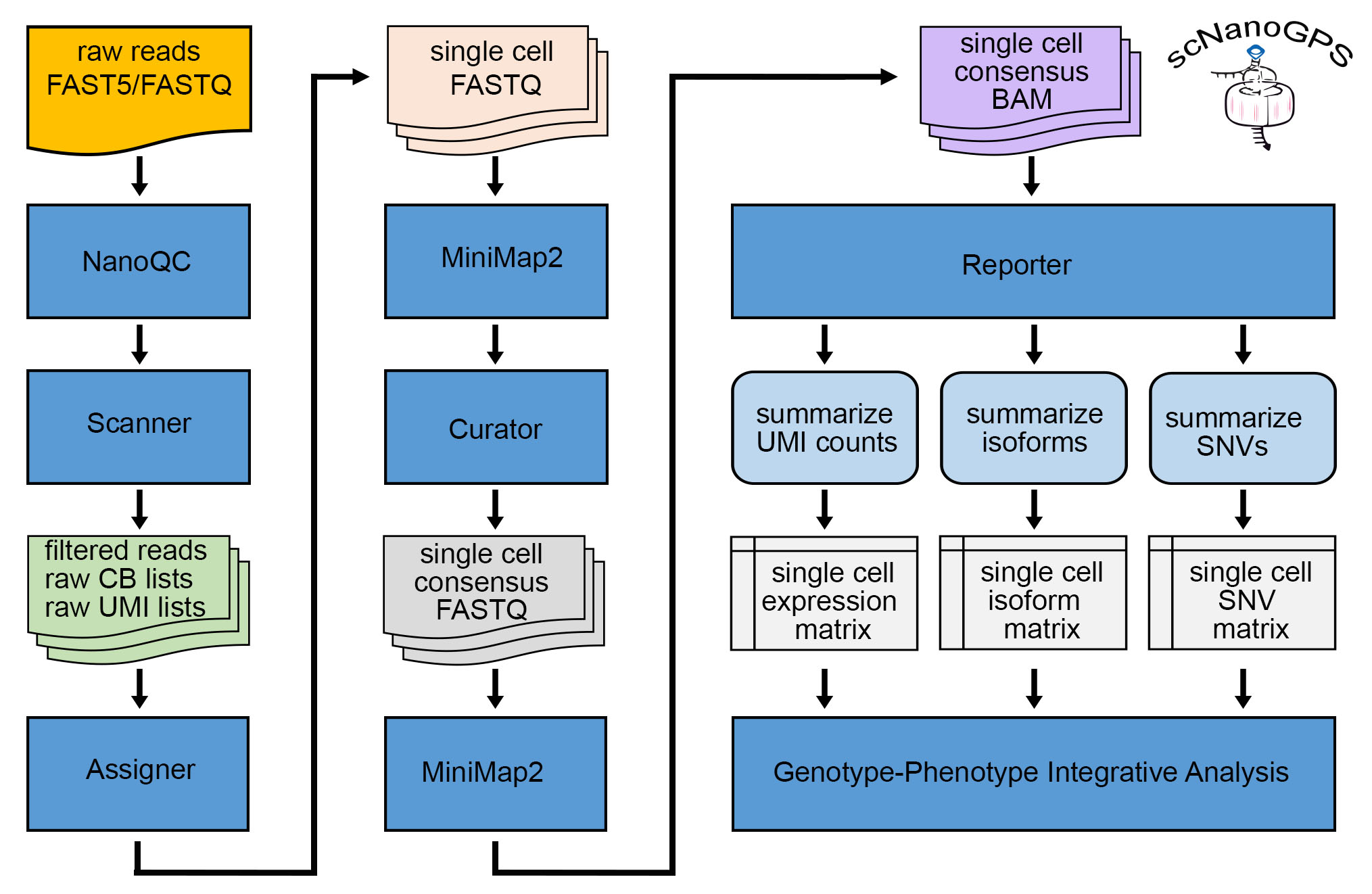
Long read single cell nanopore RNA sequencing is emerging as a powerful technology to simultaneously profile phenotypes and genotypes of same cells, which however are challenged by lacking robust computational tools and requiring paralleled short reads to curate sequencing errors. To address these challenges, we developed a computational toolkit, scNanoGPS to independently deconvolute long reads into single cells and single molecules as well as calculate both phenotypes and genotypes of same cells without paralleled short reads.
scTypeTCSingle-cell sub-typing of epithelial cells in thyroid cancer with glmnet-lasso model
Single-cell sub-typing of epithelial cells in thyroid cancer with glmnet-lasso model
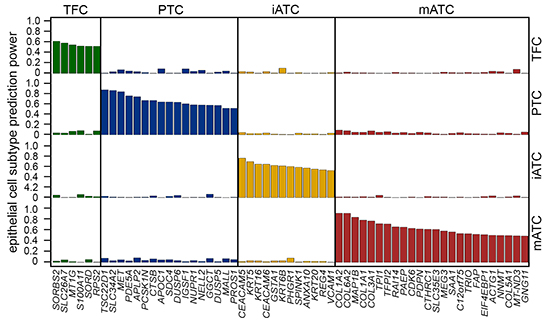
The deadliest anaplastic thyroid cancer (ATC) often transforms from indolent differentiated thyroid cancer, most commonly papillary thyroid cancer (PTC). Large-scale single-cell transcriptome data (scRNAseq) enabled the discovery of different epithelial and cancer cell subtypes occurred in different stages of thyroid cancer progression. scTypeTC is a computational tool using multinomial-lasso model to classify single cells with epithelial origins in thyroid cancer into four major subtypes, including normal thyroid follicular cells (TFCs), stress-responsive PTC cells (PTCs), inflammatory ATC cells (iATCs) and mesenchymal ATC cells (mATCs), using high-throughput scRNAseq data of normal thyroids, PTC tumors and ATC tumors. scTypeTC takes our study results as ground truth to train the model that can be used to predict new scRNAseq datasets. This vignette illustrates how to use the train model in scTypeTC to predict new single epithelial transcriptomes of thyroid tumors.
CopyKATInference of genomic copy number and subclonal structure of human tumors from high-throughput single-cell RNAseq data
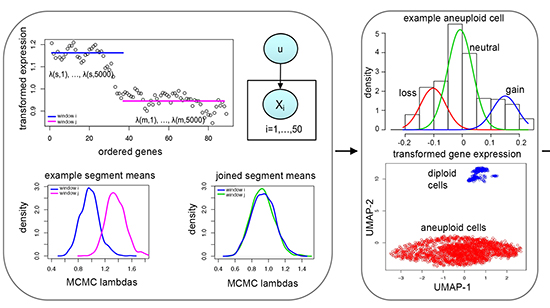
A major challenge for single-cell RNA sequencing of human tumors is to distinguish cancer cells from non-malignant cell types, as well as the presence of multiple tumor subclones. CopyKAT (Copy number Karyotyping of Tumors) is a computational tool using integrative Bayesian approaches to identify genome-wide aneuploidy at 5MB resolution in single cells to separate tumor cells from normal cells, and tumor subclones, using high-throughput sc-RNAseq data. The underlying logic for calculating DNA copy number events from RNAseq data is that gene expression levels of many adjacent genes can provide depth information to infer genomic copy number in that region. CopyKAT-estimated copy number profiles can achieve a high concordance (80 percent) with the actual DNA copy numbers obtained by whole genome DNA sequencing. The rationale for prediction tumor/normal cell states is that aneuploidy is common in human cancers (90 percent). Cells with extensive genome-wide copy number aberrations (aneuploidy) are considered as tumor cells, whereas stromal normal cells and immune cells often have 2N diploid or near-diploid copy number profiles.