Research
Opal lab research centers on understanding the pathogenesis of spinocerebellar ataxias and Giant Axonal Neuropathy, with the hope of developing viable therapies for these currently untreatable diseases.
Research on Polyglutamine Disorders: Spinocerebellar Ataxias and Related Syndromes
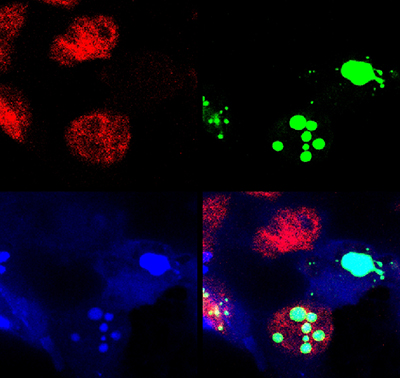 Spinocerebellar Ataxia Type 1 (SCA1), a degenerative disease that affects cerebellar Purkinje cells and brainstem neurons, is caused by a polyglutamine expansion in the involved disease protein. In this respect it is similar to agrowing number of disorders including Huntington disease that share a similar mutational mechanism. Patients with SCA1 begin to display cerebellar signs characterized by motor incoordination or ataxia in early to mid-adulthood that unfortunately progresses to more vital structures of the brain including the brainstem. Our working hypothesis is that ataxin-1 – the protein that carries the polyglutamine expansion—misfolds, is poorly cleared by the cell, and its build up causes neuronal toxicity.
Spinocerebellar Ataxia Type 1 (SCA1), a degenerative disease that affects cerebellar Purkinje cells and brainstem neurons, is caused by a polyglutamine expansion in the involved disease protein. In this respect it is similar to agrowing number of disorders including Huntington disease that share a similar mutational mechanism. Patients with SCA1 begin to display cerebellar signs characterized by motor incoordination or ataxia in early to mid-adulthood that unfortunately progresses to more vital structures of the brain including the brainstem. Our working hypothesis is that ataxin-1 – the protein that carries the polyglutamine expansion—misfolds, is poorly cleared by the cell, and its build up causes neuronal toxicity.
Developmental underpinnings of cerebellar neurodegeneration
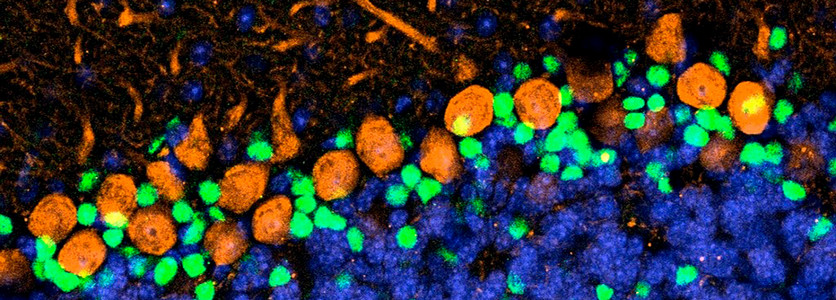
Although symptoms of ataxia in SCA1 appear relatively late in life, primarily from cerebellar dysfunction, pathogenesis begins early. Indeed, transcriptional changes detectable as early as a week after birth in SCA1-knockin mice. Given the importance of this postnatal period for cerebellar development, we have several projects aimed at understanding whether developmental processes are altered by mutant ATXN1. We have found, for instance, that expanded ATXN1 stimulates the proliferation of postnatal cerebellar stem cells in SCA1 mice. These hyperproliferating stem cells tended to differentiate into GABAergic inhibitory interneurons rather than astrocytes; this significantly increased the GABAergic inhibitory interneuron synaptic connections, disrupting cerebellar Purkinje cell function in a non–cell autonomous manner. We confirmed the increased basket cell–Purkinje cell connectivity in human SCA1 patients. Mutant ATXN1 thus alters the neural circuitry of the developing cerebellum, setting the stage for the later vulnerability of Purkinje cells to SCA1. We propose that other late-onset degenerative diseases may also be rooted in subtle developmental derailments (Edamakanti et al., Neuroscience 2018, PMID: 29533923
The Role of Inflammation in Spinocerebellar Ataxia Type 1
Most studies on SCA1 have focused on neuronal degeneration. We, however, have found that glia are impacted early in the disease. Our recent work reveals a critical role for Bergmann glia—the specialized radial glia of the cerebellum—in disease progression. Using human SCA tissue and a SCA1 mouse model, we identified a unique, JNK-dependent inflammatory pathway in Bergmann glia. Inhibiting this pathway reduced inflammation and improved disease outcomes, establishing a direct, causal role for glial dysfunction in SCA1 and highlighting a promising new therapeutic avenue for multiple SCAs (Edamakanti et al., J. Neuroinflammation 2023).
The Role of VEGF in the Pathogenesis of SCA1
We have discovered that long before the onset of clinical symptoms, mutant ATXN1 directly represses the expression of vascular endothelial growth factor (VEGF), an angiogenic cytokine with potent neurotrophic effects. This VEGF deficiency has been confirmed in autopsy samples from human SCA1 patients. Using SCA1154Q/2Q knock-in mice—a physiologically accurate model of the human disease—we discovered that reduced VEGF leads to diminished microvascular density and impaired neurotrophic support for neurons. Increasing VEGF expression, either genetically or via intracerebroventricular (ICV) delivery of recombinant VEGF, mitigates hallmark features of the disease (Cvetanovic et al., Nature Medicine 2011, PMID: 22001907). However, recombinant VEGF poses several challenges: it is biologically unstable, immunogenic, costly to manufacture, and subject to batch-to-batch variability. To overcome these limitations, we are developing fully synthetic VEGF-mimetic peptide amphiphile (PA) that engages VEGF receptors with high efficacy with a goal to developing neuroprotective therapy for SCA1 (Hu et al., Brain 2019).
Research on Giant Axonal Neuropathy (GAN)
GAN is an incurable disease affecting both the central and peripheral nervous systems. It is characterized by early-onset weakness and sensory deficits beginning in the legs, rapidly progressing to the arms and eventually affecting the rest of the body. As with all peripheral neuropathies, patients with GAN lose their deep tendon reflexes, with electrophysiological studies revealing a mixed sensory-motor axonal neuropathy. The brain, including the brainstem and cerebellum, is not spared since MRI scans show evidence of involvement of the central nervous system. At a pathological level, GAN is characterized by the presence of aggregated intermediate filaments (IF) —10 nm diameter filaments that form a network stretching from the nucleus to the cell periphery.

Deciphering neurofilament pathology and organelle failure in Giant Axonal Neuropathy
Based upon our recent findings we hypothesize that defects in gigaxonin (the protein mutated in GAN) interferes with IF degradation and clearance, ultimately filling neurons with IF bundles that interfere with axonal transport of organelles such as mitochondria to cause neuronal dysfunction and death. Current projects in the lab are aimed at testing every aspect of this hypothesis using primary neurons and genetic models of the disease.
The pathological processes involved in GAN – impaired protein degradation, IF aggregation, disorders of axonal transport and mitochondrial pathology have been described in many neurodegenerative syndromes. Thus, GAN presents a unique opportunity to study the temporal sequence of events leading specifically to IF aggregation and the relationship of these aggregates to defects in the transport of organelles. We feel that unravelling the mechanisms responsible for GAN should also provide new insights into other neurodegenerative diseases marked by IF accumulations, such as Alzheimer’s disease, Amyotrophic Lateral Sclerosis, Parkinson’s disease, Alexander disease and Neuronal Intermediate Filament Inclusion Disease.
Neurofilament accumulation is a hallmark of many neurodegenerative diseases, but in giant axonal neuropathy (GAN), it is the defining pathology. Our lab has discovered that gigaxonin, the protein mutated in this disease, is a key adaptor protein involved in neurofilament degradation. We are now studying the consequences of pathological neurofilament accumulation in this disease. Previously, we have found that neurofilament accumulations disrupt the motility of mitochondria leading to metabolic derangements. More recently, using genetic and RNA interference approaches in mouse models, we discovered that neurofilament buildup disrupts autophagy, a key degradation pathway in cells. It does so through two distinct mechanisms: by interfering with autophagosome–lysosome fusion and by mislocalizing TFEB via sequestration of the chaperone 14-3-3. These findings reveal a dual mechanism of autophagic failure that may underlie a broader spectrum of neurodegenerative disorders.
Key References:
Muhammad et al., JCI 2013, PMID: 23585478
Israeli et al., Hum Mol Genet. 2016, PMID: 27000625
Paumier et al., JCI insight 2025, PMID: 177999