Examples of Our Work
We are interested in determining how cell death occurs in age-related neurodegenerative disorders characterized by protein aggregation, such as Parkinson’s and Alzheimer’s disease. Our lab is focused on two related pathways that are critical for understanding the neurodegenerative process:
- Mechanisms that initiate and govern the conformational conversion of soluble proteins into insoluble amyloid fibrils
- The downstream neurotoxic effects of aggregated proteins with emphasis on the autophagic / lysosomal degradation system. We use analytic biochemical techniques and neurons generated from patient-derived induced pluripotent stem cells (iPSC) to study disease mechanisms of Parkinson’s disease and related amyloidoses
Culturing Patient-Derived human Neurons to Study Chronic Neurodegenerative Diseases
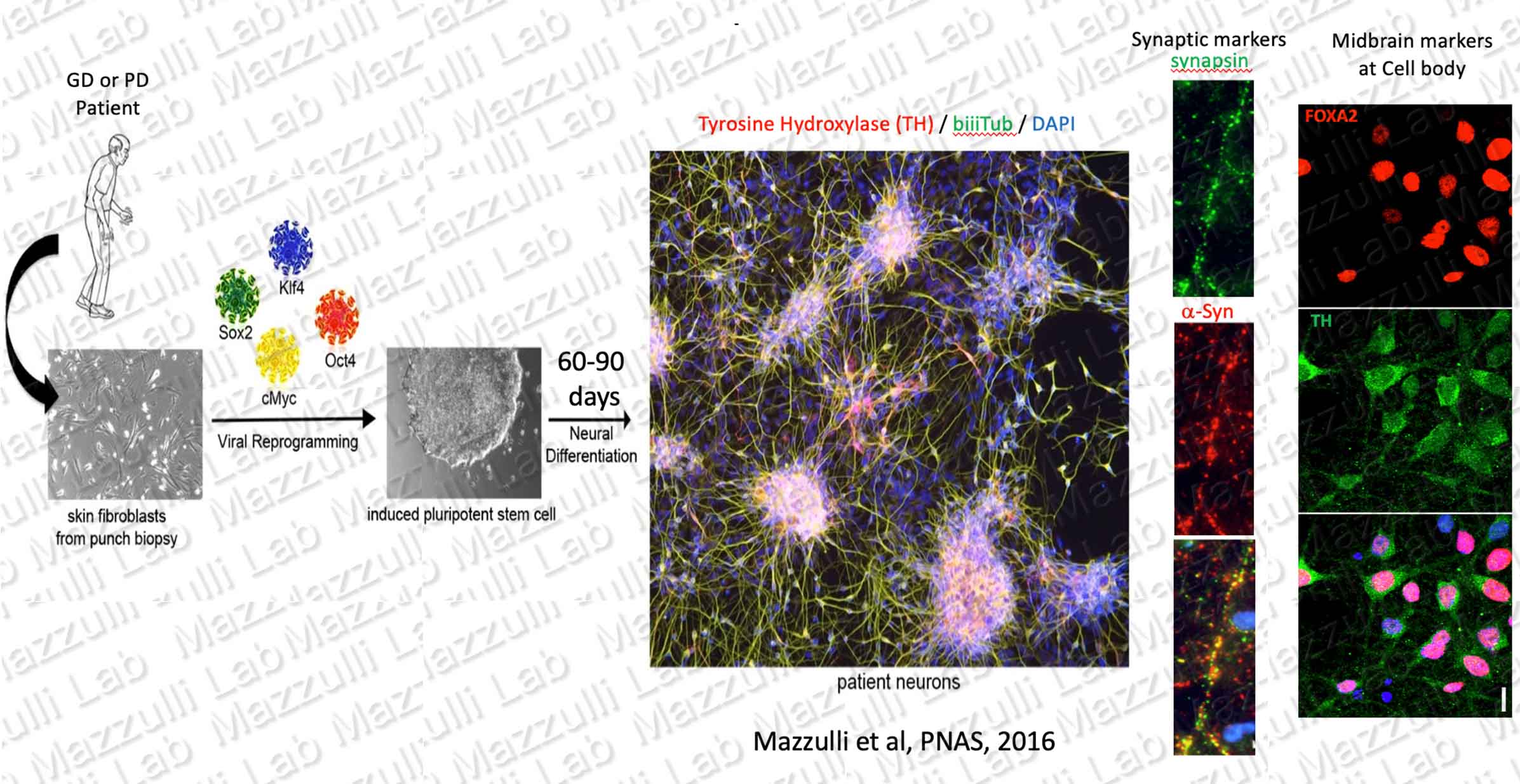
We generate induced pluripotent stem cells (iPSCs) from skin-punch fibroblast biopsies of patients with rare lysosomal and neurodegenerative disorders. iPSCs are generated by viral transduction of reprogramming factors Klf4, cMyc, Oct4, and Sox2. iPSCs are then differentiated into human neurons in vitro, providing a model to study mechanisms of amyloid formation and neurodegeneration. Neuronal marker biiiTubulin (green), midbrain maker tyrosine hydroxylase (red), and nuclei (blue) are shown in the merged image. Through careful culturing techniques, we have been able to age patient-derived neurons for hundreds of days. We use this model to study age-dependent pathological changes that occur in disease neurons, such as accumulation of a-synuclein (red) in non-synaptic locations of the neuron. A synaptic marker, synapsin, is shown in green. We published comprehensive characterizations of iPSC-derived midbrain neurons in Mazzulli et al, Cell, 2011; Mazzulli et al, PNAS 2016; Cuddy et al, Neuron 2019; and Stojkovska et al, Neuron 2022.
Protein aggregation in Neurons
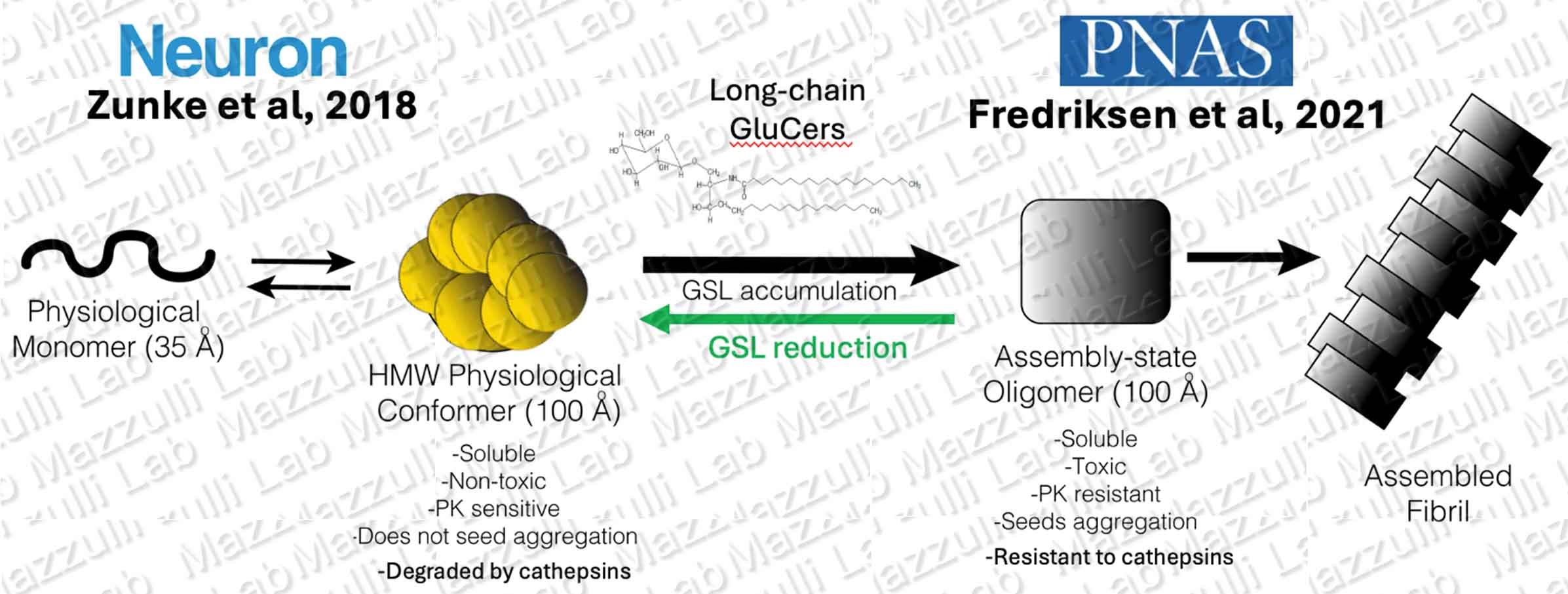
The pathways that mediate amyloid formation in the brain are not well understood. Previous studies from our lab have shown that accumulating metabolic substrates of certain lysosomal storage diseases (LSDs), such as glucosylceramide found in Gaucher disease, influence the amyloid-forming pathway of a-synuclein (Zunke et al, Neuron, 2018; Fredriksen et al, PNAS 2021). We study neurons derived from patients with LSDs to learn how lysosomal dysfunction contributes to formation of toxic protein assemblies, including amyloid fibrils and oligomeric intermediates. We also directly generate different pathogenic forms of a-synuclein made in vitro, with purified recombinant protein (electron micrograph of synthetic a-synuclein fibrils is shown on the right). Using these models, we found that glycosphingolipids (GSLs) that accumulated in Gaucher disease and GBA-PD neurons, directly convert physiological a-synuclein multimers into toxic, assembly-state oligomers that contribute to Lewy body formation (Zunke et al, Neuron, 2018). Normally, pathological protein aggregation occurs through the break-down of native protein assemblies into their monomeric components (for review, see Chiti and Dobson, Nature Chemical Biology, 2009 Jan;5(1):15-22. doi: 10.1038/nchembio.131). However our data indicates that GSLs interact and directly convert native a-syn assemblies into an alternate, toxic structure with identical hydrodynamic radius and molecular weight. GSLs with longer fatty acyl chain lengths (C22-C26) appear to be more potent at stimulating a-syn aggregation (Frediksen et al, PNAS 2021).
Proteotoxicity and Pathways to the Lysosome
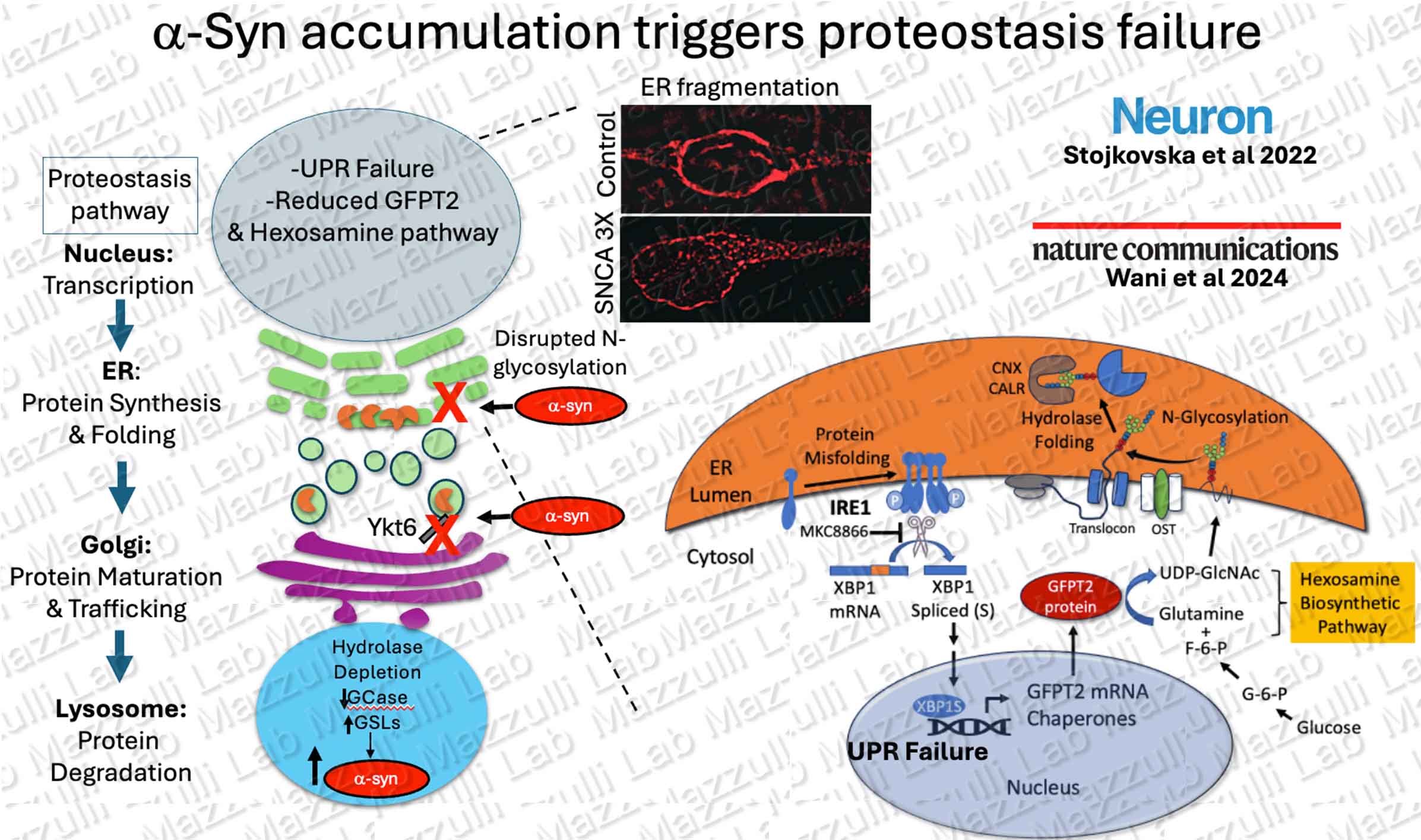
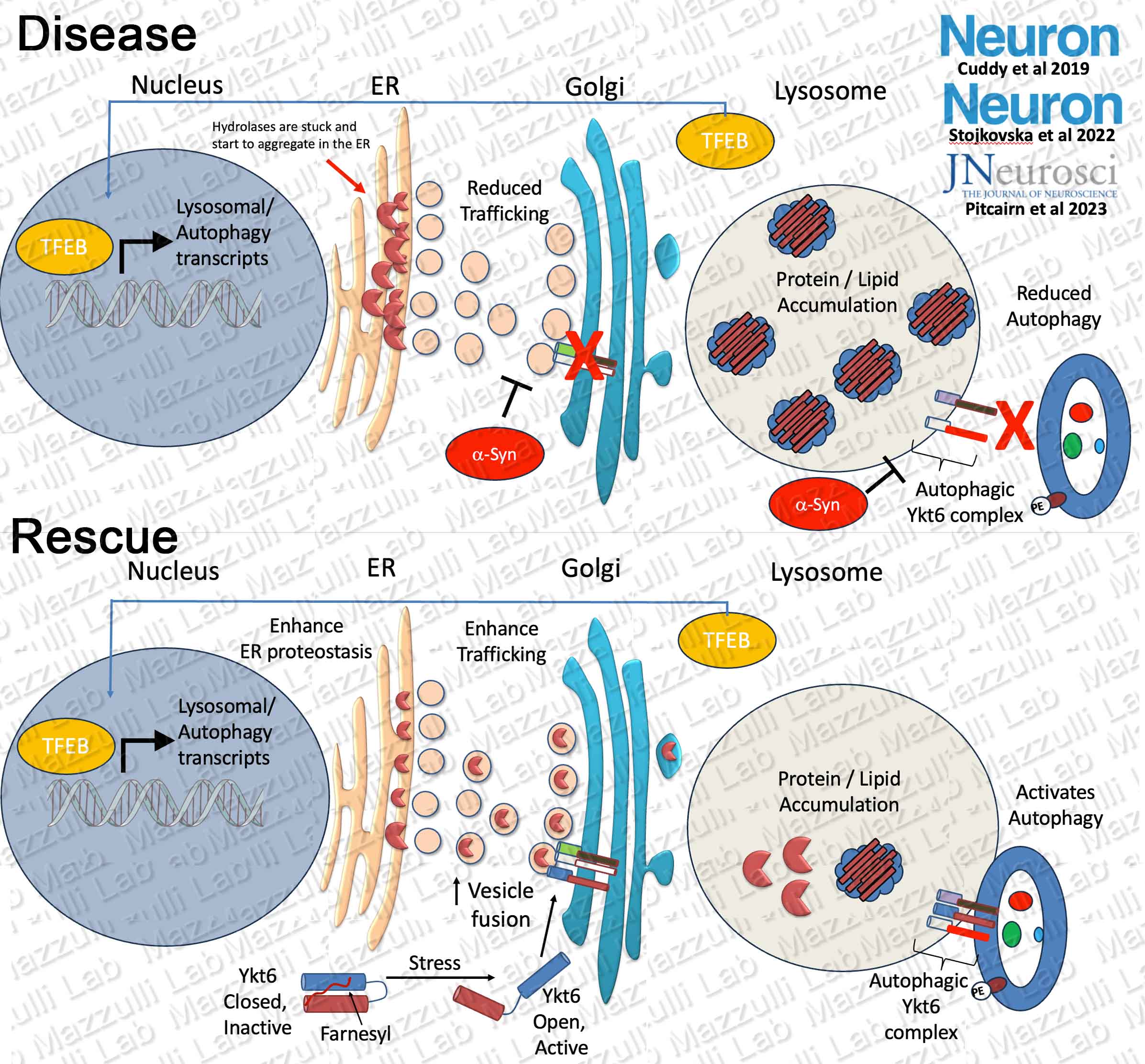
We are broadly interested in determining how protein aggregates cause cellular dysfunction, with a specific focus on the Endoplasmic Reticulum (ER) and the lysosomal degradation system. The lysosome is a critical cellular component required for recycling and degrading damaged proteins, and mediates neuronal ‘self renewal.’ The ER is responsible for maintaining protein folding and trafficking of about 1/3 of all proteins through the secretory and lysosomal pathways. We are interested in determining how macromolecular degrading enzymes mature through the secretory pathway to reach the lysosomal compartment. Our data from Parkinson patient neurons indicates that pathogenic conformations of a-synuclein disrupt ER proteostasis and downstream trafficking between the Endoplasmic Reticulum-> Golgi-> Lysosome, resulting in lysosomal enzyme depletion and dysfunction. We use Parkinson’s patient neurons that form amyloidogenic a-synuclein aggregates as a model to study how cells traffic hydrolases to the lysosome under stressful metabolic conditions. Our recent data shows that a-synuclein disrupts signaling of the unfolded protein response (UPR) between the ER and nucleus, resulting in reduced N-glycans and chaperone function (Stojkovska et al, Neuron, 2022; Wani et al, Nature Communications 2024).
---------------------------------------------------------------------------------------------------------------------------------
Translational strategies to enhance lysosomes for treatments of neurodegenerative diseases
We identify druggable biological targets in PD and DLB by studying basic disease mechanisms in human neurons. Work from our lab and others has identified perturbations in multiple branches of the proteostasis pathway in synucleinopathies including protein folding in the ER, ER-Golgi trafficking, and autophagic-lysosomal activity. We identified a biological target called Ykt6 that is capable of rescuing multiple branches of the proteostasis pathway. Ykt6 is a SNARE protein that mediates membrane fusion of ER vesicles with Golgi, and auto-lysosomal fusion. We found that ykt6 responds to physiological lysosomal stress and coordinates with the master regulation of lysosomal biogenesis, TFEB, by increasing the trafficking and supply of hydrolases into the lysosome (Cuddy et al, Neuron, 2019) and increasing autophagic-lysosomal fusion in human midbrain neurons (Pitcairn et al, J. Neurosci, 2023). Ykt6 activity is inhibited by an internal farnesyl-group, and we found that inhibiting farnesylation can activate its membrane fusing activity and enhance lysosomal function. Furthermore, we found that lysosomes can be activated even more efficiently when ykt6 activators are combined with ER proteostasis enhancers (Stojkovska et al, Neuron, 2022). We are currently testing ykt6 activators alone and in combination with other drugs that simultaneously enhance lysosomal function in patient neurons and in in vivo pre-clinical models.
---------------------------------------------------------------------------------------------------------------------------------
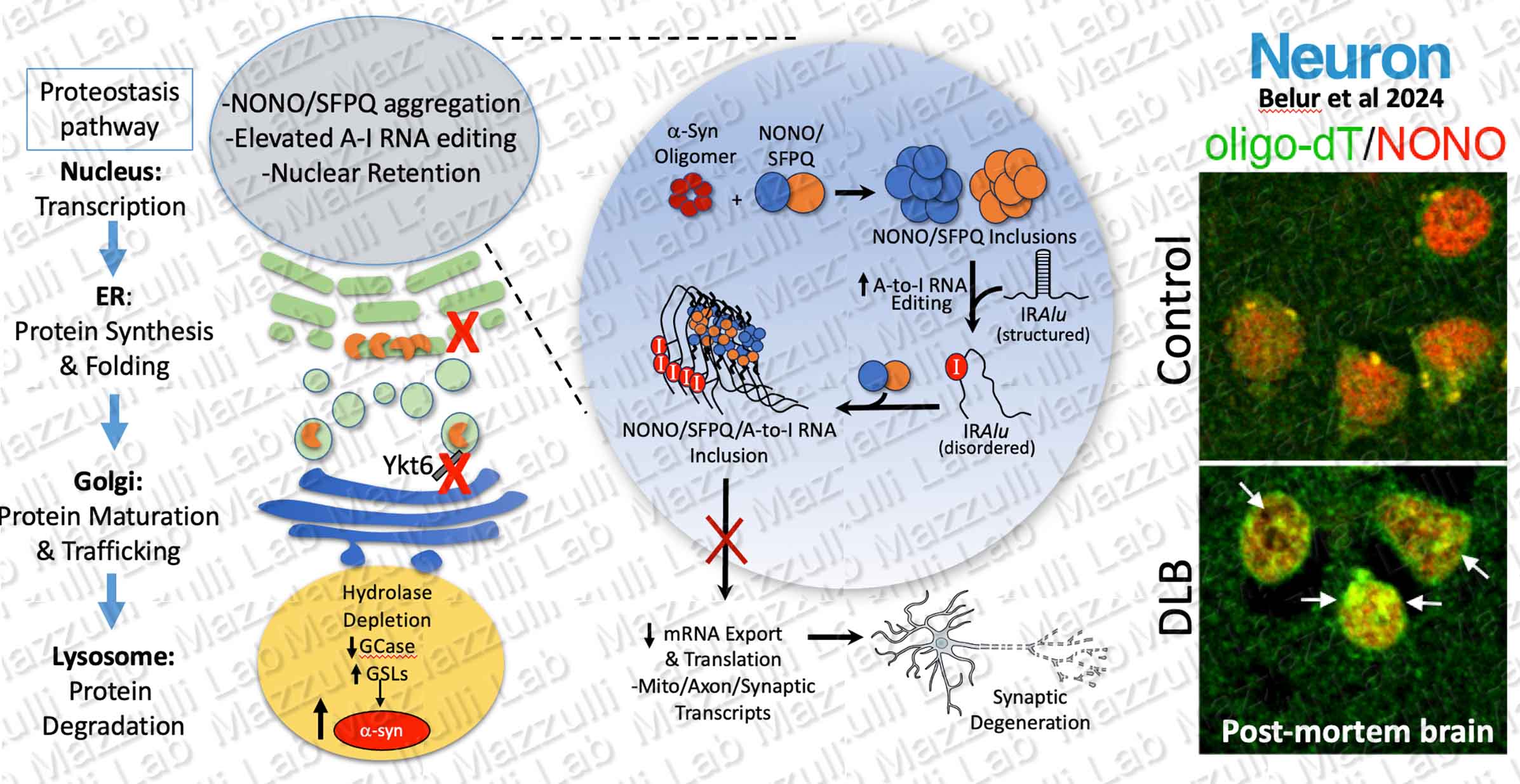
NONO/SFPQ and RNA A-to-I editing in Neurodegeneration
The majority of research in Parkinson's disease has been focused on a-synuclein aggregation. We recently discovered a novel nuclear pathology in Parkinson's disease (PD) and Dementia with Lewy body (DLB) patient brain comprised of RNA binding proteins NONO and SFPQ, and adenosine-to-inosine (A-to-I) edited RNAs (Belur et al, Neuron, 2024). Our data suggests that pathological changes in PD and DLB extend beyond a-synuclein and encompass other proteins that are susceptible to to protein aggregation such as RNA binding proteins with flexible, low complexity domains. We found that increased A-to-I editing of certain mRNAs occurs within human specific inverted repeat (IR) Alu sequences that form double stranded stem-loop structures. Introduction of inosines within Alus induces structural disorder of these RNAs and templates SFPQ inclusions by exposing SFPQ binding sites. Our lab is investigating the downstream consequences of RNA editing on neuronal health, and how editing can influence gene regulation in the nucleus of PD/DLB patients.