Role of Vimentin in NLRP3 Inflammasome Activation
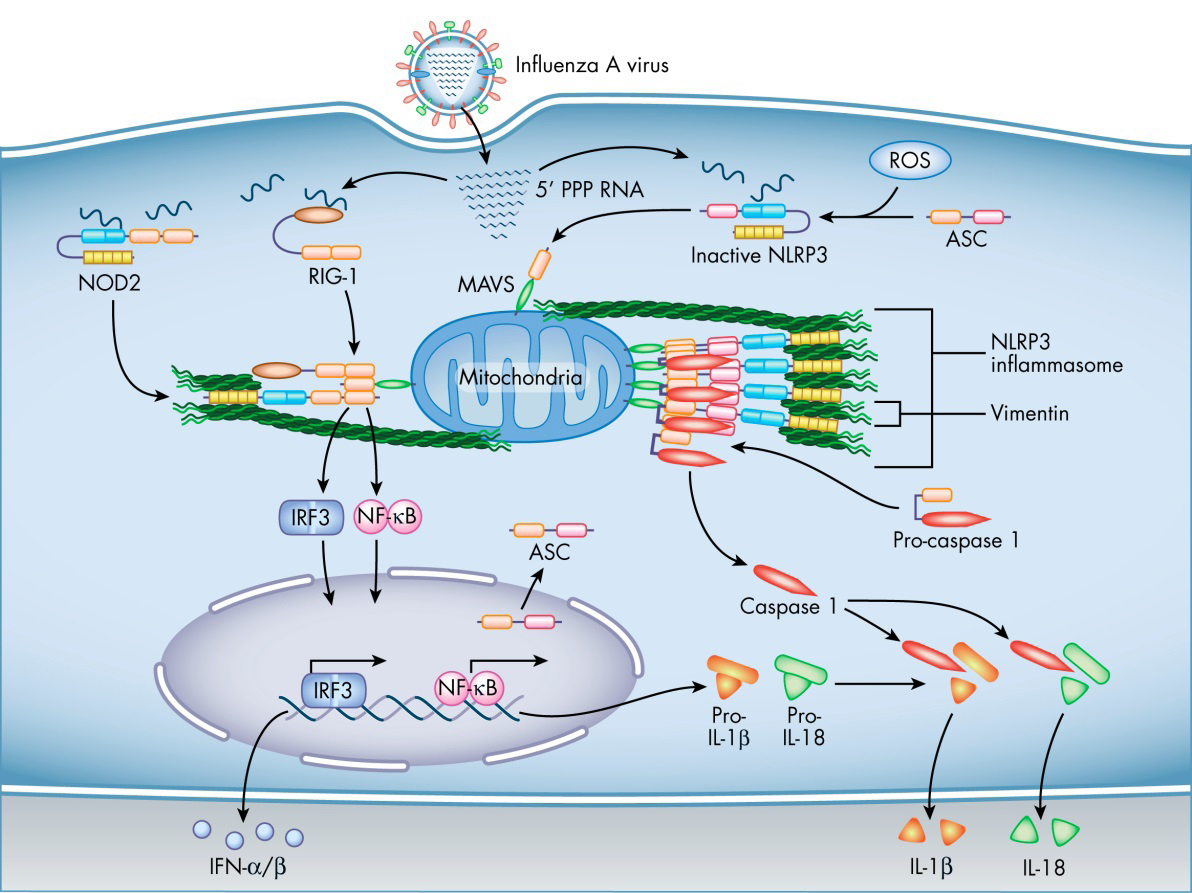
Diagram showing how vimentin may facilitate NOD2 and NLRP3 inflammasome signaling by influenza A virus.
The inflammasome is a multimolecular complex that assembles in response to pathogen- or damage-associated molecular patterns (PAMPs or DAMPs) for the production of the inflammatory cytokines IL-1β and IL-18, which recruit neutrophils to the site of infection or injury. The inflammasome consists of a pattern recognition receptor and the protease caspase-1, with the adapter molecule ASC serving to link the two [1,2].
Upon signaling from a PAMP or DAMP, the pattern recognition receptor protein undergoes a conformational change and oligomerizes, causing subsequent binding and oligomerization of ASC. The resulting molecular platform recruits the caspase-1 progenitor molecule, procaspase-1, allowing dimers to self-cleave and assemble into the enzymatically active form. Mature caspase-1 tetramers then go on to cleave the progenitors of IL-1β and IL-18 into their active forms for release from the cell [1,2].
Inflammation is necessary for the clearance of pathogens, but too much inflammation causes damage to healthy tissue. Such injury can exacerbate the severity of infectious disease. Hence, a delicate balance must be maintained to ensure the optimal host response to pathogens.
The pattern recognition receptors NLRP3 and NLRC4 are NOD-like receptors that form inflammasomes in response to infection by influenza A virus (IAV) and Pseudomonas aeruginosa, respectively [2]. We have found that activation of the NLRP3 inflammasome is regulated by the type 3 intermediate filament protein vimentin [3].
Compared with wild-type mice, vimentin-null mice infected with lethal doses of IAV show decreased production of IL-1β and IL-18, reduced lung inflammation and injury, and improved survival. We hypothesize that vimentin acts as a scaffold for the assembly of the NLRP3 and NLRC4 inflammasomes, and that its absence greatly reduces the efficiency of the activation process. Thus less IL-1β and IL-18 is produced, resulting in decreased inflammation and a corresponding reduction in disease severity.
Infection with IAV also results in activation of the NOD-like receptor NOD2, triggering phosphorylation of the transcription factor IRF3, which then translocates to the nucleus for transactivation of antiviral interferon genes [4]. NOD2 has been shown to interact with vimentin [5], suggesting that vimentin regulates the NOD2 signaling cascade during IAV infection. Hence, vimentin would be involved in both the antiviral response and the inflammatory response to IAV.
We continue to investigate the role of vimentin in inflammasome and NOD2 signaling, as well as other intersections between the antiviral and inflammatory pathways. All together, our findings contribute to greater understanding of the process of, and possible ways to counter, the damaging inflammation that can accompany severe infectious disease in the lung.
- Indramohan et al., COPs and POPs patrol inflammasome activation, J Mol Biol, 2018 (PMID: 29024695).
- dos Santos et al., The inflammasome in lung diseases, Am J Physiol Lung Cell Mol Physiol, 2012 (PMID: 22904168).
- dos Santos et al., Vimentin regulates activation of the NLRP3 inflammasome, Nat Commun, 2015 (PMID: 25762200).
- Uematsu & Akira, Toll-like receptors and type I interferons, J Biol Chem, 2007 (PMID: 17395581).
- Stevens et al., The intermediate filament protein, vimentin, is a regulator of NOD2 activity, Gut, 2013 (PMID: 22684479).