RNA Processing and Human Diseases
Overwhelming evidence supports a critical role of post-transcriptional gene regulation in the development, function and maintenance of the nervous system. We are interested in understanding post-transcriptional mechanisms regulating genes critical for neurodevelopment and neurodegeneration, including RNA binding proteins (RBPs) and non-coding RNAs.
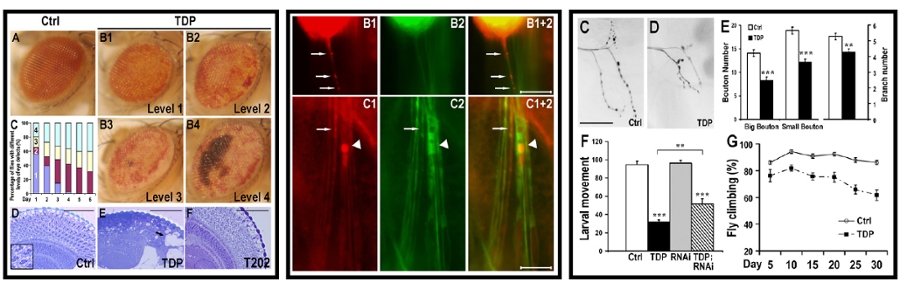
Since the discovery of TDP-43 (Tar-DNA binding protein of 43 kDa) as a characteristic component of inclusion bodies in neural tissues of patients affected by ALS (Amyotrophic lateral sclerosis) or FTLD (frontotemporal lobar degeneration), a large number of ALS-associated mutations have been identified in genes encoding RBPs, including TDP-43 and FUS. Furthermore, TDP-43 containing lesions have been found in tissue samples from patients affected by other neurodegenerative diseases, such as Alzheimer’s disease (AD) and traumatic brain injuries.
We have established cellular and animal models for TDP-43 and FUS proteinopathies (Li et al, 2010; Chen et al, 2011; Guo et al, 2011; Deng et al, 2015). We have developed multi-disciplinary approaches to examine these disease models and patient tissue samples. Using our powerful genetic models, we have begun to identify genetic modifier genes for these diseases. Using the integrated strategy, we have begun to uncover molecular and cellular defects in these neurological diseases and to understand fundamental mechanisms underlying protein aggregation and neurotoxicity associated with TDP-43 and FUS proteinopathies. Our data also suggest that mitochondrial damage may be a target in future development of diagnostic and therapeutic tools for these devastating neurodegenerative diseases.