Research
Seasonal influenza infection affects a significant proportion of the population in the US and worldwide and while most patients infected with influenza A virus (IAV) recover without sequelae, in many patients influenza virus infection may cause ARDS. Alveolar epithelial cells (AEC) are targets for IAV, and play an important role in mounting the initial host response. The Sznajder Lab hypothesizes that the alveolar epithelium plays an important effector role in protecting the lung from severe injury. Findings indicate that the degradation of PKCζ, which triggers the down-regulation of Na,K-ATPase, by the E3 ligase HOIL-1L decreases AEC death. HOIL-1L is a member of the Linear Ubiquitination Assembly Complex (LUBAC) and the lab studies whether LUBAC participates in the modulation of the inflammatory intensity in the lung epithelium during IAV infection. Also, they are investigating the mechanisms by which modest inhibition of the Na,K-ATPase, whether pharmacologic inhibition of the Na,K-ATPase by cardiotonic steroids such as ouabain are protective by inhibiting virus replication.
Proteostasis is at the center of protein function and encompasses all cellular processes that regulate protein folding, misfolding, unfolding, degradation and repair. Our studies suggest that endocrine signals from the injured lung during IAV infection can disrupt skeletal muscle proteostasis pathways and contribute to skeletal muscle dysfunction. We have found that the slower recovery of the skeletal muscle function in aged mice during IAV pneumonia is the consequence of diminished proteostatic reserve in cells responsible for regenerating the damaged skeletal muscle. Interestingly, our preliminary imaging and proteomic data suggest that signals from the lung that induce muscle degradation are counterbalanced by a robust chaperone response to preserve proteostasis in young but not old animals.
Hypercapnia (high pCO2) is observed in patients with lung diseases such as chronic obstructive pulmonary disease (COPD), cystic fibrosis, broncho-pulmonary dysplasia and advanced neuromuscular diseases. We hypothesize that hypercapnia promotes the ubiquitin-proteasome mediated degradation of diaphragmatic myofibers and impairs the function of muscle satellite cells required for its regeneration. As such, we are investigating the mechanisms by which hypercapnia leads to impaired recovery from lung injury and the effect of high CO2 on diaphragm muscle degradation and impaired regeneration.
Alveolar fluid reabsorption is effected by vectorial Na+ transport via apical Na+ channels and basolateral Na,K-ATPase of the alveolar epithelium. We and others have reported that β-adrenergic agonists upregulate the Na,K-ATPase in alveolar epithelial cells by increasing the traffic and recruitment of Na,K-ATPase containing vesicles into the cell membrane, resulting in increased catalytic activity. Moreover, GPCR-mediated upregulation of the Na,K-ATPase resulted in increased alveolar fluid clearance in normal lungs and in rodent models of lung injury. We are investigating fundamental aspects of Na,K-ATPase regulation and active Na+ transport in lungs which will help with the design of new strategies to increase lung edema clearance.
Funding
- NIA PPG Project 3: Activating proteostasis in aging resident macrophages to prevent muscle and cognitive dysfunction after pneumonia
PI: Sznajder and Misharin - NIH PPG Project 3: Targeting linear ubiquitination to attenuate inflammation and promote repair after viral pneumonia
PI: Budinger and Sznajder - NIH R01: Mitochondrial maladaptive response of the lung epithelium to elevated CO2 levels
PI: Chandel and Sznajder
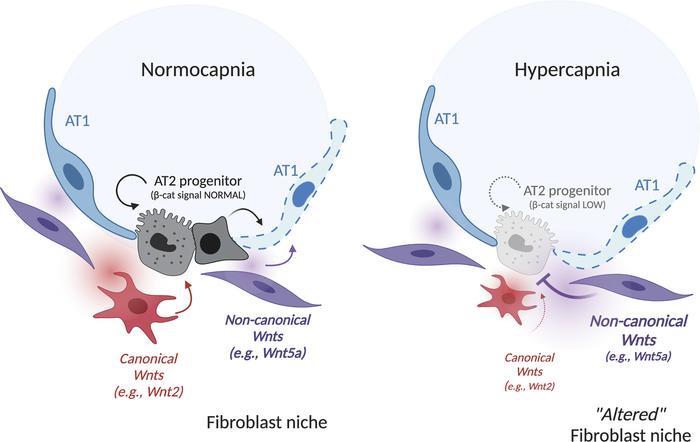
Model for hypercapnia-mediated inhibition of βcat signaling in AT2 cells via skewing of fibroblast-derived Wnts.
At the left, normocapnia is represented. AT2 progenitors are spatially proximal to Pdgfra/Wnt2-expressing fibroblasts (red/red gradient) that maintain βcat signaling and AT2 self-renewal to replace damaged AT1 cells. Pdgfra/Wnt5a-
At the right, hypercapnia leads to reduced βcat signaling in AT2 cells, impairing cell renewal and differentiation by skewing Wnt expression in PDGFRα stromal cells toward a noncanonical variety, with Wnt5a significantly elevated. Narrowness of the AT2 progenitor niche raises the possibility that elevated WNT5A release (purple gradient) in close spatial vicinity to the WNT2 signal (red gradient) antagonizes βcat signaling in AT2 cells, inhibiting proliferative capacity.
From Dada et al., JCI Insight, 2023;8(4):e159331. DOI: 10.1172/jci.insight.159331