Basic Science Research
Our lab seeks to understand the biology of neurodegenerative diseases for which there are no treatment options. We are particularly interested in genetic diseases. While relatively rare, they provide key insights into the more common disorders that afflict the brain. We hope that our research will allow us to tease out the most proximal pathological pathways that we can then target for reversal.
Research on Polyglutamine Disorders: Spinocerebellar Ataxias and Related Syndromes
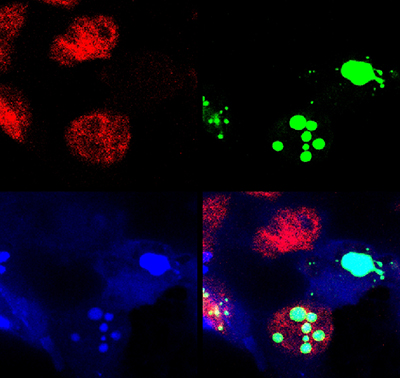
Spinocerebellar Ataxia Type 1 (SCA1), a degenerative disease that affects cerebellar Purkinje cells and brainstem neurons, is caused by a polyglutamine expansion in the involved disease protein. In this respect it is similar to agrowing number of disorders including Huntington disease that share a similar mutational mechanism. Patients with SCA1 begin to display cerebellar signs characterized by motor incoordination or ataxia in early to mid-adulthood that unfortunately progresses to more vital structures of the brain including the brainstem. Our working hypothesis is that ataxin-1 – the protein that carries the polyglutamine expansion—misfolds, is poorly cleared by the cell, and its build up causes neuronal toxicity.
Current Projects
- Transcriptional hypothesis in SCA1 pathogenesis: One of the earliest features of this disease is a change in the gene expression signature within affected neurons. We are elucidating the pattern of gene expression changes in the vulnerable Purkinje cell population and identifying the contribution of these alterations to pathology.
- The role of the vascular and angiogenic factor VEGF in SCA1 pathogenesis: One of the genes that we have already found to be down-regulated is the neurotrophic and angiogenic factor VEGF. Importantly, we have discovered that genetic or pharmacologic replenishment of VEGF mitigates SCA1 pathogenesis. These results suggest a novel therapeutic strategy for this incurable disease, as well as the possibility of cross-talk between the degenerating cerebellum and its microvasculature that we will now test experimentally. We are also extending our study more broadly to address the role of endothelial and glial pathology in SCA1.
- The role of ataxin-1 misfolding and clearance in disease pathogenesis: We are testing different strategies to decrease ataxin-1 levels and promote its clearance.
- Research on genetic parkinsonian and dystonic syndromes: In addition to work on spinocerebellar ataxia, we have also initiated projects in order to shed light on genetic parkinsonian and dystonic syndromes.
Research on Giant Axonal Neuropathy (GAN)
GAN is an incurable disease affecting both the central and peripheral nervous systems. It is characterized by early-onset weakness and sensory deficits beginning in the legs, rapidly progressing to the arms and eventually affecting the rest of the body. As with all peripheral neuropathies, patients with GAN lose their deep tendon reflexes, with electrophysiological studies revealing a mixed sensory-motor axonal neuropathy. The brain, including the brainstem and cerebellum, is not spared since MRI scans show evidence of involvement of the central nervous system. At a pathological level, GAN is characterized by the presence of aggregated intermediate filaments (IF) —10 nm diameter filaments that form a network stretching from the nucleus to the cell periphery.
In order to elucidate the pathogenesis of GAN, we have begun a collaboration with the lab of Dr. Robert Goldman, a leading expert on IF proteins. Based upon our recent findings we hypothesize that defects in gigaxonin (the protein mutated in GAN) interferes with IF degradation and clearance, ultimately filling neurons with IF bundles that interfere with axonal transport of organelles such as mitochondria to cause neuronal dysfunction and death. Current projects in the lab are aimed at testing every aspect of this hypothesis using primary neurons and genetic models of the disease.
The pathological processes involved in GAN – impaired protein degradation, IF aggregation, disorders of axonal transport and mitochondrial pathology have been described in many neurodegenerative syndromes. Thus, GAN presents a unique opportunity to study the temporal sequence of events leading specifically to IF aggregation and the relationship of these aggregates to defects in the transport of organelles. We feel that unravelling the mechanisms responsible for GAN should also provide new insights into other neurodegenerative diseases marked by IF accumulations, such as Alzheimer’s disease, Amyotrophic Lateral Sclerosis, Parkinson’s disease, Alexander disease and Neuronal Intermediate Filament Inclusion Disease. Our efforts to gain insights into GAN were initiated and facilitated by a remarkable relationship with Lori Sames, mother of Hannah, who suffers from GAN. Lori has established a GAN disease foundation called Hannah’s Hope Fund.